


细胞衰老指细胞停止生长,同时细胞形态和生理代谢活动发生显著退变的过程。主要表现在细胞生长停滞和β-半乳糖苷酶(senescence associated β-galactosidase,SA-β-gal)活化[4]。细胞衰老一方面对生物体有益,如促进伤口愈合、肿瘤抑制、血小板生成等[5]。但长期的细胞衰老和某些因素诱导的不正常细胞衰老与炎症和疾病等相关,然而机制未明[6]。已有研究证实,MIF可以调节细胞衰老,并在许多衰老相关疾病中起着重要作用[7⇓-9]。充分认识MIF在细胞衰老中的作用和机制,有助于提高对细胞衰老导致的相关疾病的认知水平,以及研发更有效的靶点药物。因此,本文就近年来MIF与细胞衰老相关的研究进展进行总结,重点是MIF调控细胞衰老的机制,为进一步认识细胞衰老机制提供帮助。
1 MIF
MIF的膜受体主要包括CD74受体,以及趋化因子受体CXCR2和CXCR4。MIF与CXCR2和CXCR4相互作用,能促进单核细胞和淋巴细胞的召集等,在炎症和肿瘤中发挥作用[1]。MIF在细胞中的作用主要是通过与CD74结合而介导,CD74与MIF结合后可以迅速内化,能触发免疫反应和肿瘤细胞增殖等一系列生物过程[17]。CD74是一种与主要组织相容性复合体Ⅱ(MHCⅡ)相关的跨膜糖蛋白,在免疫细胞中广泛表达,CD74标记约90%的低级B细胞淋巴瘤和20%的低级T细胞淋巴瘤[13]。近期一项研究发现,CD74的p41亚型可通过抑制组织蛋白酶介导的病毒蛋白质外壳裂解,对抗埃博拉病毒和新型冠状病毒(SARS-CoV-2),抑制埃博拉病毒和SARS-CoV-2进入宿主细胞[18]。如果没有CD74,MHCII无法得到正常转运,抗原提呈便会受损。感染SARS-CoV-2将诱导衰老,新冠患者的呼吸道和血清标本可检测到衰老标志物,衰老相关分泌表型SASP的水平升高[19],在体外用SARS-CoV-2菌株感染上皮Vero-E6细胞和肺泡2型(AT2)细胞,证明SARS-CoV-2感染通过激活DNA损伤和DNA损伤修复(DNA damage repair, DDR)途径诱发细胞衰老[20]。CD74具有抗病毒活性,针对CD74与病毒诱导的细胞衰老之间的关系值得研究,这是目前从单一研究衰老到在生理、病理、环境诱因复杂背景下探究衰老调控机制的大趋势。
2 细胞衰老
细胞衰老分为复制衰老(replicative senescence, RS)和压力诱导的早熟性衰老(stress-induced premature senescence, SIPS)两类。复制衰老是由于细胞不断分裂导致端粒缩短,从而激发p53信号通路引发的衰老。除了细胞内端粒缩短诱发的复制衰老以外,许多刺激性因素,如过量的氧、离子辐射均能缩短细胞的复制寿命,促进细胞衰老,这种类型的衰老称为压力诱导的早熟性衰老[21]。细胞衰老会影响许多生理和病理过程,如免疫系统疾病、癌症等[22-23]。细胞衰老的生物学特征是细胞周期停滞[24],即永久停止生长,此过程涉及两条信号通路:p53/p21/RB和p16INK4A/RB。这两条信号通路会控制细胞周期,并决定细胞是否分裂[25]。另外,有研究证明自噬也与细胞衰老密切相关,细胞自噬功能下降会导致细胞衰老[26-27]。
3 MIF与细胞衰老的关系
3.1 MIF延缓细胞衰老的机制
3.1.1 MIF通过抑制衰老基因表达延缓细胞衰老
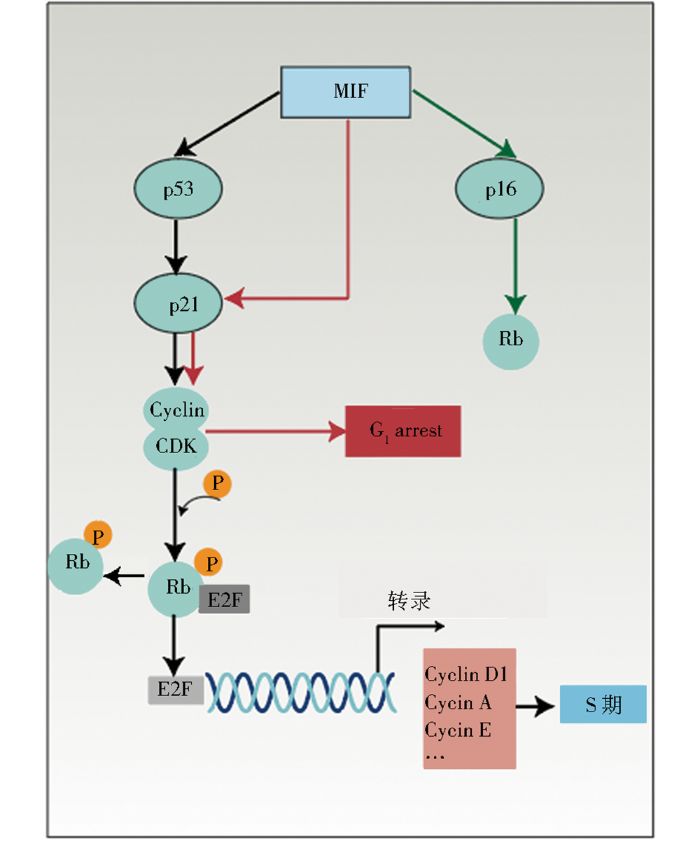
细胞衰老的特点是细胞周期停滞,细胞周期停滞信号主要位于p53/p21/RB和p16INK4A/RB通路。MIF能够通过上述通路调控细胞周期(图1),在细胞周期中,特别在G1/S转变期间,通过细胞周期蛋白依赖性激酶(cyclin-dependent kinase, CDK)磷酸化的RB与E2F分离,并允许 E2F 介导的S期促进因子,如细胞周期蛋白(cyclin)的转录,细胞才被允许由G1期向S期进行转换。若RB磷酸化水平降低,则E2F转录因子的后续作用将受到阻碍,导致细胞分裂停止,从而诱发衰老。MIF与RB-E2F信号通路密切相关,首先,MIF基因缺失的细胞表现出E2F依赖性生长改变,MIF基因缺失将使成纤维细胞增殖能力降低,而增加E2F的表达可以恢复成纤维细胞的增殖能力,即MIF通过依赖E2F的机制影响细胞增殖[36]。其次,MIF可能通过调节RB-E2F通路的活性影响肿瘤生长,早期研究发现,抑制MIF的mRNA表达,小鼠的结肠肿瘤变小,且在体外培养结肠癌细胞时发现,抑制MIF,RB和E2F的转录活性都受到抑制[37]。MIF对p21基因的调控也会影响细胞周期。例如,抑制MIF表达将会上调肌原细胞内p21的水平,从而出现G1期细胞积累的现象[38]。也会导致宫颈腺癌细胞的细胞周期停滞[39]。反之,MIF过表达,p21表达减少,细胞周期进程也将改变。例如,在大鼠气道平滑肌细胞中,增加MIF表达会导致p21表达下降,cyclin D1、cyclin D3及CDK6表达上升,引起S/G2期细胞的积累[40]。
图1
3.1.2 MIF过表达延缓细胞衰老的机制
图2
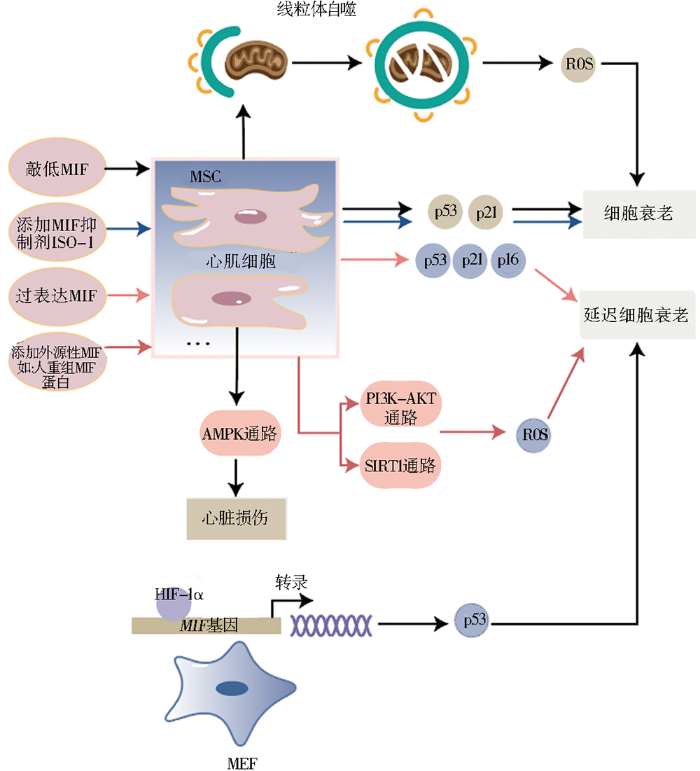
外源性MIF的增加也能够对抗自然老化和物理、化学刺激诱导的细胞衰老。正常衰老的SD大鼠BM-MSCs与年轻SD大鼠相比,其细胞生物学功能受损,表现在增殖能力减弱、信号转导功能降低以及对外界刺激的抗性下降等。向衰老BM-MSCs加入重组MIF培养,细胞的生物学功能恢复显著,细胞增殖率和一些作为营养指标的蛋白质表达水平均增高[43]。阿霉素诱导SD大鼠BM-MSCs衰老,添加外源性MIF后可通过PI3K-AKT信号通路和抑制氧化应激,使衰老的BM-MSCs恢复活力,细胞增殖率和端粒酶活性均显著增加[8]。用携带MIF的外泌体处理细胞,发现携带MIF的外泌体能够通过LncRNA-NEAT1/miR-221-3p/SIRT2 信号通路,抵抗阿霉素诱导的心脏衰老[44]。向辐射诱导衰老的人心肌细胞中添加人重组MIF继续培养,细胞增殖能力较未添加MIF的衰老心肌细胞强,细胞周期调控因子的表达水平降低,SA-β-gal衰老阳性细胞百分比也显著下降。这个对抗衰老的过程通过MIF靶向SIRT1抑制miR-34a实现。此研究表明添加外源性MIF能对抗辐射诱导的细胞衰老[9]。
3.1.3 敲低MIF将促进细胞衰老
从年轻捐献者骨髓中分离出MSCs,通过基因敲除降低细胞中MIF表达,相关研究显示,衰老标志基因p53和p21的表达显著升高,衰老细胞数量也显著增加[7]。衰老会降低心脏抗压能力,增加心脏相关疾病的易感性。老年人心脏中的MIF水平与年轻人相比下调,且AMPK活性降低。研究表明敲除MIF会通过抑制AMPK信号通路的活化增加心脏损伤,证明因AMPK信号通路介导的反应受损引发的心脏相关疾病,可能是由MIF表达水平降低导致的[45]。体外衰老模型显示,MIF基因缺失加剧了阿霉素诱导的H9C2成肌细胞早衰,但对细胞中p16和p21的蛋白质表达无影响,而是通过影响自噬加剧细胞老化[31],如图2所示。说明MIF也可能通过影响自噬而不是通过触发p53/p21/RB、p16INK4A/RB通路调控细胞衰老。
3.2 氧化应激反应在MIF调控细胞衰老中的作用和机制
活性氧(reactive oxygen species, ROS)是细胞衰老的常见介质,是含氧的化学物质,包括过氧化物、羟自由基、超氧阴离子等。ROS是氧的正常代谢副产物,但在高温、紫外线等环境压力下,ROS水平会急剧增加,机体内氧化与抗氧化作用平衡失调,将对细胞造成严重损伤,这一过程即氧化应激。氧化应激是细胞衰老的重要诱因[46]。
MIF与氧化应激过程密切相关。一方面,研究表明抗氧化反应原件的激活对保护细胞免受氧化应激至关重要[47],而MIF在抗氧化反应原件介导的基因调控中起着关键作用[48]。MIF已被证明可以保护细胞免受氧化应激诱导的细胞凋亡,氧化应激反应导致细胞内MIF浓度升高,MIF浓度升高会通过调节细胞内谷胱甘肽浓度抑制细胞凋亡[15,49]。另一方面,MIF具有氧化还原活性,可以减少氧化应激反应,MIF基因缺失的心脏成纤维细胞暴露于氧化应激条件下,细胞内ROS增加2.3倍,而过表达MIF后,细胞内ROS恢复原来水平[50]。研究证实,MIF基因缺失通过抑制线粒体自噬增加了人髓核细胞中ROS的积累和衰老相关标志物的表达,加剧人髓核细胞衰老[27]。另外,ROS反过来也可诱导MIF分泌。研究者已证实在乳腺癌细胞系中,抑制自噬,细胞内ROS分泌增加,触发MIF分泌[51-52]。
3.3 HIF-1α在MIF调控细胞衰老中的作用和机制
当氧气需求超过供应量时,局部组织或整个身体中的氧气水平下降,称为缺氧。缺氧可诱导衰老,也可导致衰老过程中功能的下降。
缺氧反应由缺氧诱导因子(hypoxia-inducible factor 1alpha, HIF)-1α控制[53]。实验证明,MIF与HIF-1α相关,在小鼠的胚胎成纤维细胞(murine embryonic fibroblasts, MEFs)中,MIF是HIF-1α的关键效应因子,HIF-1α延缓细胞衰老需要MIF的存在(图2),MIF是HIF-1α和衰老之间的潜在联系,通过p53发挥作用[28]。HIF-1α能够调节MIF表达,缺氧条件下,血管平滑肌细胞中HIF-1α活化,刺激MIF表达,敲低HIF-1α后,MIF基因和蛋白质的表达受抑制[54]。在巨噬细胞中,缺氧会诱导HIF-1α活化,激活MIF,促进巨噬细胞杀灭细菌,但HIF-1α调节MIF产生的机制尚不清楚[55]。研究表明,MIF存在于鼠囊肿细胞中并促进囊肿生长,在鼠囊肿细胞中,HIF-1α特异性敲除阻止了MIF的上调和囊肿生长,此过程还包括cAMP的参与[56]。反过来,MIF也可以调节HIF-1α表达。肺癌细胞中,MIF通过NF-κB调节HIF-1α[57]。对伤口未愈合病人脂肪干细胞样本的研究表明,MIF和其受体CD74在缺氧条件下显著升高,重组MIF刺激了HIF-1α的表达,从而调节炎症因子如IL-1β,影响伤口环境[58]。在乳腺癌细胞中,MIF以p53依赖的方式调节HIF-1α的活性[59]。
近年来将MIF和HIF-1α联系在一起针对细胞衰老的研究并不多,大多数研究倾向于炎症方向,MIF在炎症与衰老通路之间的调节机制也许更为复杂,有待进一步阐明。
4 MIF功能多态性与衰老相关疾病
MIF 调节多种衰老相关疾病,包括神经退行性疾病、癌症、心血管疾病、呼吸系统疾病等[62⇓⇓-65],既有不利影响也有有利作用。MIF对阿尔茨海默病、帕金森病及肌萎缩侧索硬化等神经退行性疾病具有保护作用,肌萎缩侧索硬化是由SOD1突变导致线粒体功能障碍引起的,MIF通过抑制SOD1突变和促凋亡线粒体途径的活化发挥保护作用[62]。在帕金森病中,MIF可以增加抗炎因子,减少促炎因子,抑制炎症反应。也可以降低聚腺苷二磷酸核糖聚合酶1(poly ADP-ribose polymerase 1, PARP1)裂解,下调促凋亡基因(Bax)表达,抑制细胞凋亡[66]。阿尔茨海默病的典型特征是淀粉样蛋白β蛋白积累诱发的慢性炎症,MIF可以直接与淀粉样蛋白β蛋白相互作用,保护神经细胞免受毒害[67]。在癌症中,MIF通过p53途径干扰细胞周期[68],并激活MAPK信号通路刺激肿瘤细胞生长和血管内皮生长因子表达,对免疫系统造成负面影响,为肿瘤生长和扩散创造有利环境[69]。此外,MIF还可以增加肿瘤细胞的侵袭和耐药性[70]。MIF是动脉粥样硬化形成的关键介质,当MIF与趋化因子受体CXCR2和CXCR4结合时,将促进动脉粥样硬化白细胞募集和炎症[71],但与CD74结合又将激活AMPK和ERK信号通路保护心脏[72]。阻断MIF可能成为消除动脉粥样硬化斑块的一种策略[71],但由于MIF功能的复杂性,此方法迟迟未进入临床试验阶段。临床研究发现,在肺炎、急性呼吸窘迫综合征、慢性阻塞性肺疾病、特发性肺纤维化等年龄相关性肺病患者的组织和细胞中,MIF含量升高[64]。但在动物实验中,抑制MIF的表达有双重作用。一方面,抑制MIF表达能保护小鼠免受脂多糖、大肠杆菌感染等因素诱导的急性呼吸窘迫综合征的影响;另一方面,MIF-/-小鼠较正常小鼠更易受到肺部损伤,这与MIF的抗氧化、抑制DNA损伤等功能有关[73-74]。
MIF还参与由衰老导致的人体器官衰退而引起的疾病,如耳聋、骨关节炎。老年性耳聋与MIF有关,老年MIF敲除鼠听觉丧失程度增加,且伴随内耳畸形[75]。实验证明,MIF基因缺失也可以降低老年小鼠骨关节炎的严重程度[76]。但也有研究发现骨关节炎患者血浆中MIF的mRNA和蛋白质水平较对照组显著降低[77],因此目前对于MIF在骨关节炎中的作用还存在争议。由于老年人体内局部胶原蛋白、成纤维细胞和巨噬细胞减少,因此老年人组织修复能力差,伤口愈合慢。雌激素是伤口愈合过程的主要调节剂,可以改变与年龄相关的伤口愈合过程,MIF参与伤口愈合过程。研究发现,人类伤口中MIF水平随着年龄的增长而增加,且受到雌激素下调[78]。
5 MIF生物工程应用
鉴于MIF及其受体参与调控炎症、癌症、免疫系统疾病等多种疾病机制,以MIF为靶标治疗疾病的思路早已被采用并进行了大量研究。基于MIF及其受体为中心研发的生物工程药物已进入临床试验阶段,主要有抗MIF抗体、抗CD74抗体、抗CXCR4抗体以及一些小分子抑制剂。
Imalumab是一种全人源的单克隆抗体,能够与氧化型MIF特异性结合干扰细胞存活和增殖的关键信号通路,如ERK1/2和AKT信号通路,激活细胞中的胱天蛋白酶-3抑制前列腺癌细胞增殖,促进细胞凋亡[79-80]。Milatuzumab是一种靶向CD74的新型免疫治疗药物,其在多发性骨髓瘤细胞系中显示出抗增殖活性,研究发现,Milatuzumab通过干扰CD74活性,阻碍NF-κB信号通路激活,导致细胞生长被抑制,使多发性骨髓瘤细胞株凋亡[81]。Ulocuplumab是首创全人源IgG4 单克隆抗CXCR4抗体,能通过阻断CXCR4与CXCL12结合诱导肿瘤细胞凋亡,抑制肿瘤细胞增殖、细胞扩散和骨髓内外迁移等过程[82],应用于巨球蛋白血症、慢性淋巴细胞白血病和多发性骨髓瘤等疾病中[83⇓-85]。MEDI3185也是一种抗CXCR4抗体,通过阻止CXCR4与趋化因子基质衍生因子-1的结合阻断CXCR4信号转导,抑制肿瘤生长[86]。
小分子抑制剂指能够靶向作用于蛋白质,降低蛋白质活性或阻碍生化反应的、分子量小于1 kDa的化合物,MIF的小分子抑制剂有ISO-1、NAPQI、4-IPP等[87]。ISO-1能竞争性的与MIF底物催化位点结合,抑制MIF活性,能改善炎症,抑制癌细胞增殖、迁移和肿瘤生长[88-89]。而NAPQI、4-IPP通过与MIF共价结合改变MIF结合其他分子的能力,抑制MIF发挥作用[90]。此外,Repertaxin、P425等小分子抑制剂通过抑制MIF与其受体的结合阻断MIF的活性,对炎症、自身免疫疾病、癌症等疾病具有治疗效果[91-92]。国内首款针对MIF研发的小分子抑制剂IPG1094已于2021年8月在美国启动临床试验。
以上生物工程药物靶向MIF及其受体,是治疗疾病的有效策略之一,在MIF相关复杂疾病中广泛应用。然而诸如此类药物在临床试验过程也会产生不良反应和副作用[93]。纳米抗体是大羊驼自然产生的微小抗体,是一种有效、新颖的制剂,参与疾病治疗,以其体积小、组织渗透性强、低免疫原性、高溶解度等优势成为生物制剂的候选者[94]。近期研究发现,纳米抗体可以高效对抗新冠病毒[95-96],可成为治疗新冠感染的新手段。有研究表明,抗MIF纳米体能阻断MIF的互变异构酶活性,抑制炎症,减轻脓毒性休克的致死性[97]。此外,首例MIF降解剂MD13于2021年设计合成,MD13利用蛋白质水解靶向嵌合体技术彻底消除MIF,并消除蛋白质-蛋白质相互作用,可诱导MIF在A549和HEK293细胞系中降解,有效降低MIF在细胞中的蛋白质水平,诱导G2/M期细胞周期阻滞,抑制细胞增殖[98]。
6 总结与展望
以上研究说明,MIF基因过表达和增加人重组MIF均可延缓细胞衰老,无论自然衰老的细胞或是诱导衰老的细胞。MIF能够通过抑制氧化应激延缓衰老,也可能与HIF-1α的相互调节调控细胞衰老。衰老的细胞能被MIF改善,表现在衰老相关基因的表达降低、衰老细胞数量减少、细胞增殖率增加等方面;反之,降低MIF的表达,细胞会呈现衰老加剧趋势,出现细胞中衰老相关基因表达上调、衰老细胞数量显著升高、细胞自噬异常和细胞周期停滞等现象。因此,本文认为MIF的浓度与细胞衰老密切相关,MIF浓度降低,导致细胞功能下降,细胞生理活动异常,通过一系列反应激活p53/p21/RB、p16INK4A/RB通路,或影响自噬触发细胞衰老。当MIF浓度恢复,细胞才发挥正确功能,进行正常生理代谢活动。
针对MIF调控细胞衰老的功能,我们可以利用MIF延缓正常细胞衰老,为抑制细胞衰老提供新的思路,甚至通过MIF调控细胞周期来触发肿瘤细胞分裂周期停滞,以达到治疗疾病的目的。此外,受MIF调控的多种疾病与衰老密切相关,其中疾病与MIF复杂的关联等着被探索,基于MIF为靶点的抗衰老药物也有待研发。
参考文献
Evolving complexity of MIF signaling

Macrophage migration inhibitory factor (MIF) is a cytokine expressed in various cell types, including hematopoietic, epithelial, endothelial, mesenchymal and neuronal cells. Altered MIF expression has been associated with a multitude of diseases ranging from inflammatory disorders like sepsis, lupus and rheumatoid arthritis to organ pathologies such as heart failure, myocardial infarction, acute kidney injury, organ fibrosis and a number of malignancies. The implication of MIF in these diseases was supported by numerous animal studies. MIF acts in an autocrine and paracrine manner via binding and activating the receptors CD74/CD44, CXCR2, CXCR4 and CXCR7. Upon receptor binding, several downstream signaling pathways were shown to be activated in vivo, including ERK1/2, AMPK and AKT. Expression of MIF receptors is not uniform in various cells, resulting in differential responses to MIF across various tissues and pathologies. Within cells, MIF can directly bind and interact with intracellular proteins, such as the constitutive photomorphogenic-9 (COP9) signalosome subunit 5 (CSN5), p53 or thioredoxin-interacting protein (TXNIP). D-dopachrome tautomerase (D-DT or MIF-2) was recognized to be a structural and functional homolog of MIF, which could exert overlapping effects, raising further the complexity of canonical MIF signaling pathways. Here, we provide an overview of the expression and regulation of MIF, D-DT and their receptors. We also discuss the downstream signaling pathways regulated by MIF/D-DT and their pathological roles in different tissue, particularly in the heart and the kidney.Copyright © 2019 Elsevier Inc. All rights reserved.
Rediscovering MIF: new tricks for an old cytokine
Produced by many cell types, macrophage migration inhibitory factor (MIF) is a pleiotropic cytokine with critical and supporting roles in many disease states and conditions. Its disease associations, myriad functions, receptors, and downstream signaling have been the subject of considerable research, yet many questions remain. Moreover, the relevance of MIF's partially functionally redundant family member, D-dopachrome tautomerase (D-DT), also remains to be further characterized. Here, we discuss recent discoveries demonstrating direct roles of MIF in supporting NLR Family Pyrin Domain-Containing 3 (NRLP3) inflammasome activation, as well as acting as a molecular chaperone for intracellular proteins. These findings may offer new clues to understanding MIF's multiple functions, and assist the development of putative MIF-targeting therapeutics for a variety of pathologies.Copyright © 2019 Elsevier Ltd. All rights reserved.
Macrophage migration inhibitory factor exerts pro-proliferative and anti-apoptotic effects via CD 74 in murine hepatocellular carcinoma
What is and what is not cell senescence
Cell senescence is a process that occurs due to telomere erosion or can be induced by various stresses. Senescent cells cease to divide but remain alive, metabolically active and able to secrete many molecules. They also show many hallmarks of senescence, such as enlarged size, increased granularity, increased activity of SA-β-galactosidase, increased level of cyclin-dependent kinase inhibitors, p16 and p21, and DNA damage foci. Originally, cell senescence was attributed to proliferating normal cells, in contrast to cancer cells, which were considered as those endowed with indefinite growth ability. Recently, it has become evident that anticancer treatment induces senescence in cancer cells. Moreover, certain hallmarks of senescence were detected in non-proliferating post-mitotic cells. There are many signalling pathways involved in cell senescence, but the most prevalent is the DNA damage response pathway. In this review we have summarized our long lasting input in the global study of the mechanisms of senescence of normal and cancer cells and discussed the diversity of the concept of cell senescence.
The role of cellular senescence in tissue repair and regeneration
Cellular senescence: a view throughout organismal life
Cellular senescence is the final fate of most cells in response to specific stimuli, but is not the end. Indeed, it is the beginning of a singular life, with multiple side roads leading to diverse effects on the organism. Many studies have been done in the last few years to elucidate the intriguing role of senescent cells in the organism, demonstrating them as the cause of several age-related diseases. However, these cells are also positively implicated in other important pathways, such as embryogenesis and wound healing. It appears that the multiple effects are time-dependent: long-term senescence is mostly implicated in chronic inflammation and disease, whereas in the short term, senescent cells seem to be beneficial, being rapidly targeted by the innate immune system. The influence of senescent cells on their neighbors by paracrine factors, differential activity depending on developmental stage, and duration of the effects make the cellular senescent program a unique spatial-temporal mechanism. During pathological conditions such as progeroid syndromes, this mechanism is deregulated, leading to accelerated onset of some aging-related diseases and a shorter lifespan, among other physiological defects. Here, we review the three primary cell senescence programs described so far (replicative, stress-induced, and developmentally programmed senescence), their onset during development, and their potential roles in diseases with premature aging. Finally, we discuss the role of immune cells in keeping senescence burden below the threshold of disease.
Macrophage migration inhibitory factor rejuvenates aged human mesenchymal stem cells and improves myocardial repair
Macrophage migration inhibitory factor rescues mesenchymal stem cells from doxorubicin-induced senescence though the PI3K-Akt signaling pathway
Doxorubicin (DOXO), an anthracycline antibiotic, is a commonly used anticancer drug. Despite its widespread usage, the therapeutic effects of DOXO are limited by its cardiotoxicity. Mesenchymal stem cell (MSC)-based therapies have had positive outcomes in the treatment of DOXO-induced cardiac damage; however, DOXO exerts toxic effects on MSCs, decreasing the effectiveness of MSC therapy. Macrophage migration inhibitory factor (MIF) promotes MSC survival and rejuvenation, and thus is a promising candidate to protect MSCs against DOXO-induced injury. The present study revealed that DOXO induced the senescence of MSCs, resulting in decreased proliferation, viability and paracrine effects. However, pretreatment with MIF improved the proliferation rate, viability, paracrine function, telomere length and telomerase activity of MSCs. Furthermore, the results indicated that the molecular mechanism underlying the anti-senescent function of MIF involved the phosphatidylinositol 3-kinase (PI3K)-RAC-α serine/threonine-protein kinase (Akt) signaling pathway, which MIF activated. In agreement with this finding, silencing Akt was identified to abolish the anti-senescent effect of MIF. In addition, MIF decreased oxidative stress in MSCs, as revealed by the decreased production of reactive oxygen species and malondialdehyde, and the increased activity of superoxide dismutase. These results indicate that MIF can rescue MSCs from a state of DOXO-induced senescence by inhibiting oxidative stress and activating the PI3K-Akt signaling pathway. Thus, treatment with MIF may have an important therapeutic application for the rejuvenation of MSCs in patients with cancer being treated with DOXO.
Macrophage migration inhibitory factor serves a pivotal role in the regulation of radiation-induced cardiac senescencethrough rebalancing the microRNA-34a/sirtuin 1 signaling pathway
Role of macrophage migration inhibition factor in kidney disease
Macrophage migration inhibitory factor (MIF) has been shown to play a pathogenic role in kidney disease. This article will review the current understanding of the expression of MIF and its functional role in immune-mediated renal injury in both human and animal models of kidney disease. Upregulation of MIF is found in both human and experimental kidney disease including renal allograft rejection and contributes significantly to macrophage and T-cell accumulation and progressive renal injury. It is now clear that MIF is a stress factor, a pro-inflammatory cytokine, a growth factor and a hormone. MIF acts through many mechanisms to mediate renal injury including the innate and adaptive immune systems, the induction of cytokines, chemokines, adhesion molecules as well as interactions with glucocorticoids and the hypothalamic-pituitary-adrenal axis. MIF exerts its biological activities via signaling through its CD74/CD44 receptor complex to activate the downstream ERK1/2 MAP kinase. The functional importance of MIF in kidney disease is demonstrated by the findings that treatment with a neutralizing anti-MIF antibody is able to prevent or reverse renal injury in crescentic anti-GBM glomerulonephritis. In addition, mice null for MIF are protected against immune-mediated lupus nephritis. MIF plays a critical role in kidney diseases and further studies of the functional role and signaling mechanisms of MIF in human kidney diseases are needed.
Machine learning in medicine
Spurred by advances in processing power, memory, storage, and an unprecedented wealth of data, computers are being asked to tackle increasingly complex learning tasks, often with astonishing success. Computers have now mastered a popular variant of poker, learned the laws of physics from experimental data, and become experts in video games - tasks that would have been deemed impossible not too long ago. In parallel, the number of companies centered on applying complex data analysis to varying industries has exploded, and it is thus unsurprising that some analytic companies are turning attention to problems in health care. The purpose of this review is to explore what problems in medicine might benefit from such learning approaches and use examples from the literature to introduce basic concepts in machine learning. It is important to note that seemingly large enough medical data sets and adequate learning algorithms have been available for many decades, and yet, although there are thousands of papers applying machine learning algorithms to medical data, very few have contributed meaningfully to clinical care. This lack of impact stands in stark contrast to the enormous relevance of machine learning to many other industries. Thus, part of my effort will be to identify what obstacles there may be to changing the practice of medicine through statistical learning approaches, and discuss how these might be overcome. © 2015 American Heart Association, Inc.
Macrophage migration inhibitory factor regulates integrin-β1 and cyclin D 1 expression via ERK pathway in podocytes
The biological function and significance of CD 74 in immune diseases
Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1
The cytokine macrophage migration inhibitory factor reduces pro-oxidative stress-induced apoptosis
The cytokine macrophage migration inhibitory factor (MIF) exhibits pro- and anti-inflammatory activities and regulates cell proliferation and survival. We investigated the effects of MIF on apoptosis. As MIF exhibits oxidoreductase activity and participates in regulating oxidative cell stress, we studied whether MIF could affect oxidative stress-induced apoptosis. We demonstrated that MIF exhibits antiapoptotic activity in various settings. MIF suppressed camptothecin-induced apoptosis in HeLa and Kym cells and HL-60 promyeloblasts. Both exogenous MIF and endogenous MIF, induced following overexpression through tetracycline (tet) gene induction, led to significant suppression of apoptosis. Apoptosis reduction by MIF was also observed in T cells. A role for MIF in redox stress-induced apoptosis was addressed by comparing the effects of rMIF with those of the oxidoreductase mutant C60SMIF. Endogenous overexpression of C60SMIF was similar to that of MIF, but C60SMIF did not suppress apoptosis. Exogenous rC60SMIF inhibited apoptosis. A role for MIF in oxidative stress-induced apoptosis was directly studied in HL-60 leukocytes and tet-regulated HeLa cells following thiol starvation or diamide treatment. MIF protected these cells from redox stress-induced apoptosis and enhanced cellular glutathione levels. As overexpressed C60SMIF did not protect tet-regulated HeLa cells from thiol starvation-induced apoptosis, it seems that the redox motif of MIF is important for this function. Finally, overexpression of MIF inhibited phosphorylation of endogenous c-Jun induced by thiol starvation, indicating that MIF-based suppression of apoptosis is mediated through modulation of c-Jun N-terminal kinase activity. Our findings show that MIF has potent antiapoptotic activities and suggest that MIF is a modulator of pro-oxidative stress-induced apoptosis.
Role of macrophage migration inhibitory factor in heat-induced apoptosis in keratinocytes
The multifaceted roles of the invariant chain CD74:more than just a chaperone
The invariant chain (CD74) is well known for its essential role in antigen presentation by mediating assembly and subcellular trafficking of the MHCII complex. Beyond this, CD74 has also been implicated in a number of processes independent of MHCII. These include the regulation of endosomal trafficking, cell migration and cellular signalling as surface receptor of the pro-inflammatory cytokine macrophage migration inhibitory factor (MIF). In several forms of cancer, CD74 is up-regulated and associated with enhanced proliferation and metastatic potential. In this review, an overview of the diverse biological functions of the CD74 protein is provided with a particular focus on how these may be regulated. In particular, proteolysis of CD74 will be discussed as a central mechanism to control the actions of this important protein at different levels. Copyright © 2016 Elsevier B.V. All rights reserved.
MHC class II transactivator CIITA induces cell resistance to Ebola virus and SARS-like coronaviruses
Recent outbreaks of Ebola virus (EBOV) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have exposed our limited therapeutic options for such diseases and our poor understanding of the cellular mechanisms that block viral infections. Using a transposon-mediated gene-activation screen in human cells, we identify that the major histocompatibility complex (MHC) class II transactivator (CIITA) has antiviral activity against EBOV. CIITA induces resistance by activating expression of the p41 isoform of invariant chain CD74, which inhibits viral entry by blocking cathepsin-mediated processing of the Ebola glycoprotein. We further show that CD74 p41 can block the endosomal entry pathway of coronaviruses, including SARS-CoV-2. These data therefore implicate CIITA and CD74 in host defense against a range of viruses, and they identify an additional function of these proteins beyond their canonical roles in antigen presentation.Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Virus-induced senescence is a driver and therapeutic target in COVID-19
Pulmonary infection by SARS-CoV-2 induces senescence accompanied by an inflammatory phenotype in severe COVID-19: possible implications for viral mutagenesis
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection of the respiratory system can progress to a multisystemic disease with aberrant inflammatory response. Cellular senescence promotes chronic inflammation, named senescence-associated secretory phenotype (SASP). We investigated whether coronavirus disease 2019 (COVID-19) is associated with cellular senescence and SASP.
Cellular senescence: molecular mechanisms and pathogenicity
Cellular senescence is the arrest of normal cell division. Oncogenic genes and oxidative stress, which cause genomic DNA damage and generation of reactive oxygen species, lead to cellular senescence. The senescence-associated secretory phenotype is a distinct feature of senescence. Senescence is normally involved in the embryonic development. Senescent cells can communicate with immune cells to invoke an immune response. Senescence emerges during the aging process in several tissues and organs. In fact, increasing evidence shows that cellular senescence is implicated in aging-related diseases, such as nonalcoholic fatty liver disease, obesity and diabetes, pulmonary hypertension, and tumorigenesis. Cellular senescence can also be induced by microbial infection. During cellular senescence, several signaling pathways, including those of p53, nuclear factor-κB (NF-κB), mammalian target of rapamycin, and transforming growth factor-beta, play important roles. Accumulation of senescent cells can trigger chronic inflammation, which may contribute to the pathological changes in the elderly. Given the variety of deleterious effects caused by cellular senescence in humans, strategies have been proposed to control senescence. In this review, we will focus on recent studies to provide a brief introduction to cellular senescence, including associated signaling pathways and pathology.© 2018 Wiley Periodicals, Inc.
Cellular senescence in aging and age-related disease: from mechanisms to therapy
Cellular senescence, a process that imposes permanent proliferative arrest on cells in response to various stressors, has emerged as a potentially important contributor to aging and age-related disease, and it is an attractive target for therapeutic exploitation. A wealth of information about senescence in cultured cells has been acquired over the past half century; however, senescence in living organisms is poorly understood, largely because of technical limitations relating to the identification and characterization of senescent cells in tissues and organs. Furthermore, newly recognized beneficial signaling functions of senescence suggest that indiscriminately targeting senescent cells or modulating their secretome for anti-aging therapy may have negative consequences. Here we discuss current progress and challenges in understanding the stressors that induce senescence in vivo, the cell types that are prone to senesce, and the autocrine and paracrine properties of senescent cells in the contexts of aging and age-related diseases as well as disease therapy.
Cell senescence and fibrotic lung diseases
Hallmarks of cellular senescence
Cellular senescence is a permanent state of cell cycle arrest that promotes tissue remodeling during development and after injury, but can also contribute to the decline of the regenerative potential and function of tissues, to inflammation, and to tumorigenesis in aged organisms. Therefore, the identification, characterization, and pharmacological elimination of senescent cells have gained attention in the field of aging research. However, the nonspecificity of current senescence markers and the existence of different senescence programs strongly limit these tasks. Here, we describe the molecular regulators of senescence phenotypes and how they are used for identifying senescent cells in vitro and in vivo. We also highlight the importance that these levels of regulations have in the development of therapeutic targets.Copyright © 2018 Elsevier Ltd. All rights reserved.
Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype
Cellular senescence is a stable cell cycle arrest that can be triggered in normal cells in response to various intrinsic and extrinsic stimuli, as well as developmental signals. Senescence is considered to be a highly dynamic, multi-step process, during which the properties of senescent cells continuously evolve and diversify in a context dependent manner. It is associated with multiple cellular and molecular changes and distinct phenotypic alterations, including a stable proliferation arrest unresponsive to mitogenic stimuli. Senescent cells remain viable, have alterations in metabolic activity and undergo dramatic changes in gene expression and develop a complex senescence-associated secretory phenotype. Cellular senescence can compromise tissue repair and regeneration, thereby contributing toward aging. Removal of senescent cells can attenuate age-related tissue dysfunction and extend health span. Senescence can also act as a potent anti-tumor mechanism, by preventing proliferation of potentially cancerous cells. It is a cellular program which acts as a double-edged sword, with both beneficial and detrimental effects on the health of the organism, and considered to be an example of evolutionary antagonistic pleiotropy. Activation of the p53/p21WAF1/CIP1and p16INK4A/pRB tumor suppressor pathways play a central role in regulating senescence. Several other pathways have recently been implicated in mediating senescence and the senescent phenotype. Herein we review the molecular mechanisms that underlie cellular senescence and the senescence associated growth arrest with a particular focus on why cells stop dividing, the stability of the growth arrest, the hypersecretory phenotype and how the different pathways are all integrated.
Autophagy and senescence: a new insight in selected human diseases
Senescence and autophagy play important roles in homeostasis. Cellular senescence and autophagy commonly cause several degenerative processes, including oxidative stress, DNA damage, telomere shortening, and oncogenic stress; hence, both events are known to be interrelated. Autophagy is well known for its disruptive effect on human diseases, and it is currently proposed to have a direct effect on triggering senescence and quiescence. However, it is yet to be proven whether autophagy has a positive or negative impact on senescence. It is known that elevated levels of autophagy induce cell death, whereas inadequate autophagy can trigger cellular senescence. Both have important roles in human diseases such as aging, renal degeneration, neurodegenerative disorders, and cancer. Therefore, this review aims to highlight the relevance of senescence and autophagy in selected human ailments through a summary of recent findings on the connection and effects of autophagy and senescence in these diseases.© 2019 Wiley Periodicals, Inc.
Deficiency of MIF accentuates overloaded compression-induced nucleus pulposus cell oxidative damage via depressing mitophagy
HIF1α delays premature senescence through the activation of MIF
Premature senescence in vitro has been attributed to oxidative stress leading to a DNA damage response. In the absence of oxidative damage that occurs at atmospheric oxygen levels, proliferation of untransformed cells continues for extended periods of time. We have investigated the role of the hypoxia-inducible factor 1α (HIF1α) transcription factor in preventing senescence in aerobic and hypoxic conditions. Using embryonic fibroblasts from a conditional HIF1α knockout mouse, we found that loss of HIF1α under aerobic conditions significantly accelerated the onset of cellular senescence, and decreased proliferation under hypoxia. Furthermore, we identify the macrophage migration inhibitory factor (MIF) as a crucial effector of HIF1α that delays senescence. Inhibition of MIF phenocopies loss of HIF1α. Our findings highlight a novel role for HIF1α under aerobic conditions, and identify MIF as a target responsible for this function.
Macrophage migration inhibitory factor (MIF) promotes cell survival and proliferation of neural stem/progenitor cells
In a previous study, we showed that murine dendritic cells (DCs) can increase the number of neural stem/progenitor cells (NSPCs) in vitro and in vivo. In the present study, we identified macrophage migration inhibitory factor (MIF) as a novel factor that can support the proliferation and/or survival of NSPCs in vitro. MIF is secreted by DCs and NSPCs, and its function in the normal brain remains largely unknown. It was previously shown that in macrophages, MIF binds to a CD74-CD44 complex. In the present study, we observed the expression of MIF receptors in mouse ganglionic-eminence-derived neurospheres using flow cytometry in vitro. We also found CD74 expression in the ganglionic eminence of E14 mouse brains, suggesting that MIF plays a physiological role in vivo. MIF increased the number of primary and secondary neurospheres. By contrast, retrovirally expressed MIF shRNA and MIF inhibitor (ISO-1) suppressed primary and secondary neurosphere formation, as well as cell proliferation. In the neurospheres, MIF knockdown by shRNA increased caspase 3/7 activity, and MIF increased the phosphorylation of Akt, Erk, AMPK and Stat3 (Ser727), as well as expression of Hes3 and Egfr, the products of which are known to support cell survival, proliferation and/or maintenance of NSPCs. MIF also acted as a chemoattractant for NSPCs. These results show that MIF can induce NSPC proliferation and maintenance by multiple signaling pathways acting synergistically, and it may be a potential therapeutic factor, capable of activating NSPC, for the treatment of degenerative brain disorders.
Macrophage migration inhibitory factor regulates AKT signaling in hypoxic culture to modulate senescence of human mesenchymal stem cells
Hypoxic culture has been shown to delay premature senescence occurring during in vitro culture. Human mesenchymal stem cells (hMSCs) cultured under hypoxia have been reported to maintain their stemness properties and delay senescence compared to the cells cultured under normoxia. However, the molecular mechanism by which hypoxia regulates premature senescence has not been fully revealed. In this study, hMSCs were cultured under the conditions of 21% (normoxia) and 1% O2 (hypoxia) tension and analyzed for cell growth, expression of MSC surface markers, multilineage differentiation, and cellular senescence. Our results showed that more cells retained MSC surface markers in hypoxic culture than those in normoxic culture, and hypoxia was able to enhance multilineage differentiation of hMSCs. The hypoxic condition also delayed cellular senescence of hMSCs, increased activation of AKT signaling, and upregulated both intra- and extracellular levels of macrophage migration inhibitory factor (MIF) compared to the normoxic condition. Inhibition of AKT activity in hypoxic culture increased the number of cells with positive staining for senescence-associated β-galactosidase activity, upregulated expression levels of senescence-associated markers p16 and p21 mRNA transcripts, and decreased expression levels of potency-associated markers, including NANOG, OCT3/4, and SOX2. On the other hand, upregulated intra- and extracellular levels of MIF by stable MIF overexpression in normoxic culture increased the activation of AKT while decreasing mRNA expression of senescence-associated markers and increasing expression of potency-associated markers. Taken together, our findings suggest that hMSCs in hypoxic culture produce endogenous MIF to activate AKT signaling to delay the progression of cellular senescence.
Macrophage migration inhibitory factor (MIF) deficiency exacerbates aging-induced cardiac remodeling and dysfunction despite improved inflammation: role of autophagy regulation
Aging leads to unfavorable geometric and functional sequelae in the heart. The proinflammatory cytokine macrophage migration inhibitory factor (MIF) plays a role in the maintenance of cardiac homeostasis under stress conditions although its impact in cardiac aging remains elusive. This study was designed to evaluate the role of MIF in aging-induced cardiac anomalies and the underlying mechanism involved. Cardiac geometry, contractile and intracellular Ca2+ properties were examined in young (3-4 mo) or old (24 mo) wild type and MIF knockout (MIF-/-) mice. Our data revealed that MIF knockout exacerbated aging-induced unfavorable structural and functional changes in the heart. The detrimental effect of MIF knockout was associated with accentuated loss in cardiac autophagy with aging. Aging promoted cardiac inflammation, the effect was attenuated by MIF knockout. Intriguingly, aging-induced unfavorable responses were reversed by treatment with the autophagy inducer rapamycin, with improved myocardial ATP availability in aged WT and MIF-/- mice. Using an in vitro model of senescence, MIF knockdown exacerbated doxorubicin-induced premature senescence in H9C2 myoblasts, the effect was ablated by MIF replenishment. Our data indicated that MIF knockout exacerbates aging-induced cardiac remodeling and functional anomalies despite improved inflammation, probably through attenuating loss of autophagy and ATP availability in the heart.
Aging of the cells: insight into cellular senescence and detection Methods
MIF maintains the tumorigenic capacity of brain tumor-initiating cells by directly inhibiting p53
Tumor-initiating cells thought to drive brain cancer are embedded in a complex heterogeneous histology. In this study, we isolated primary cells from 21 human brain tumor specimens to establish cell lines with high tumorigenic potential and to identify the molecules enabling this capability. The morphology, sphere-forming ability upon expansion, and differentiation potential of all cell lines were indistinguishable in vitro However, testing for tumorigenicity revealed two distinct cell types, brain tumor-initiating cells (BTIC) and non-BTIC. We found that macrophage migration inhibitory factor (MIF) was highly expressed in BTIC compared with non-BTIC. MIF bound directly to both wild-type and mutant p53 but regulated p53-dependent cell growth by different mechanisms, depending on glioma cell line and p53 status. MIF physically interacted with wild-type p53 in the nucleus and inhibited its transcription-dependent functions. In contrast, MIF bound to mutant p53 in the cytoplasm and abrogated transcription-independent induction of apoptosis. Furthermore, MIF knockdown inhibited BTIC-induced tumor formation in a mouse xenograft model, leading to increased overall survival. Collectively, our findings suggest that MIF regulates BTIC function through direct, intracellular inhibition of p53, shedding light on the molecular mechanisms underlying the tumorigenicity of certain malignant brain cells. Cancer Res; 76(9); 2813-23. ©2016 AACR.©2016 American Association for Cancer Research.
Role of lincRNA-p21 in the protective effect of macrophage inhibition factor against hypoxia/serum deprivation-induced apoptosis in mesenchymal stem cells
Macrophage migration inhibitory factor deficiency in chronic obstructive pulmonary disease
The pathogenesis of chronic obstructive pulmonary disease (COPD) remains poorly understood. Cellular senescence and apoptosis contribute to the development of COPD; however, crucial regulators of these underlying mechanisms remain unknown. Macrophage migration inhibitory factor (MIF) is a pleiotropic cytokine that antagonizes both apoptosis and premature senescence and may be important in the pathogenesis of COPD. This study examines the role of MIF in the pathogenesis of COPD. Mice deficient in MIF (Mif −/−) or the MIF receptor CD74 (Cd74 −/−) and wild-type (WT) controls were aged for 6 mo. Both Mif −/− and Cd74 −/− mice developed spontaneous emphysema by 6 mo of age compared with WT mice as measured by lung volume and chord length. This was associated with activation of the senescent pathway markers p53/21 and p16. Following exposure to cigarette smoke, Mif −/− mice were more susceptible to the development of COPD and apoptosis compared with WT mice. MIF plasma concentrations were measured in a cohort of 224 human participants. Within a subgroup of older current and former smokers (n = 72), MIF concentrations were significantly lower in those with COPD [8.8, 95%CI (6.7–11.0)] compared with those who did not exhibit COPD [12.7 ng/ml, 95%CI (10.6–14.8)]. Our results suggest that both MIF and the MIF receptor CD74 are required for maintenance of normal alveolar structure in mice and that decreases in MIF are associated with COPD in human subjects.
Macrophage migration inhibitory factor MIF interferes with the Rb-E2F pathway
Macrophage migration inhibitory factor (MIF) is implicated in the regulation of inflammation and cell growth. We previously showed that MIF is a potent modulator of p53- and E2F-dependent pathways that are activated in response to oncogenic signaling. Here, we characterize the functional link between MIF and E2F transcription factors. Our results demonstrate that MIF-deficient cells exhibit E2F-dependent growth alterations and reduced susceptibility to oncogenic transformation. The basis for this transformation resistance is a perturbed function of the C-terminal Rb binding region of E2F4. However, inactivation of Rb or substitution of the E2F4 C-terminal domain by the E2F1 C-terminal region rescues the transformation defect. Importantly, the involvement of E2F factors in DNA replication rather than in regulation of transcription determines their oncogenic properties in the context of MIF deficiency. A proinflammatory molecule interfering with tumor suppression and DNA replication provides a compelling molecular link for the association of chronic inflammation and tumorigenesis.
Suppression of tumor growth through introduction of an antisense plasmid of macrophage migration inhibitory factor
The potential role in cell growth of Macrophage migration inhibitory factor (MIF) has been studied, however, the mechanism of its anti-tumor effect is poorly understood. Antisense-MIF plasmids were directly injected into colon 26 tumors embedded in the back of mice. Furthermore, the role of MIF in the cell cycle was assessed with regard to retinoblastoma (Rb) protein and transcription factor E2F. Plasmids containing sense- and antisense-MIF genes were transfected into human colon cancer KM12SM cells in vitro. To examine the Rb protein-E2F pathway, plasmids containing each specific cis-acting enhancer for Rb protein and E2F with luciferase reporter genes, pRB-luc and pE2F-luc, respectively, were used. Antisense MIF treatment significantly reduced the tumor size. In vitro cell proliferation was significantly suppressed by the antisense treatment as examined by BrdU uptake. Transcriptions of Rb protein were 8.4x10(3) (RLU), 9.5x10(3) and 24.3x10(3) in the antisense MIF, PBK, and the sense MIF, respectively. As for E2F, transcription activities were 3.8x10(3), 3.6x10(3) and 7.7x10(3), respectively. These results indicate the possibility that MIF may promote tumor growth, in which the activation-inactivation mechanism of the Rb protein-E2F pathway could be profoundly involved.
Macrophage migration inhibitory factor in the regulation of myoblast proliferation and differentiation
Obesity is documented to be a state of chronic mild inflammation associated with increased macrophage infiltration into adipose tissue and liver and skeletal muscle. As a pleiotropic inflammatory mediator, macrophage migration inhibitory factor (MIF) is associated with metabolic disease, so MIF may signal molecular links between adipocytes and myocytes. MIF expression was modified during myoblast differentiation, but the role of MIF during this process is unclear. C2C12 cells were transfected with MIF to investigate their role during differentiation. MIF expression attenuated C2C12 differentiation. It did not change proliferation, but downregulated cyclin D1 and CDK4, causing cell accumulation in the G1 phase. p21 protein was increased significantly and MyoD, MyoG, and p21 mRNA also increased significantly in the C2C12 cells treated with ISO-1, suggesting that inhibition of MIF promotes differentiation. MIF inhibits the myoblast differentiation by affecting the cell cycle progression, but does not affect proliferation.
Macrophage immigration inhibitory factor promotes cell proliferation and inhibits apoptosis of cervical adenocarcinoma
Macrophage migration inhibitory factor (MIF) promotes rat airway muscle cell proliferation and migration mediated by ERK1/2 and FAK signaling
Macrophage migration inhibitory factor (MIF) is an inflammatory mediator that contributes to asthmatic airway remodeling; however, little is known regarding the effects of MIF on airway smooth muscle cells (ASMCs). In the present study, we found that an enhanced expression of MIF promoted ASMC proliferation, increased the population of cells in the S/G2 phase, downregulated P21 expression, and upregulated cyclin D1, cyclin D3, and Cdk6 expression. In addition, the apoptosis of ASMCs was significantly decreased in response to MIF overexpression, compared with the negative control. Moreover, MIF facilitated the migration of ASMCs by upregulating the expression of matrix metalloproteinase (MMP)-2. Finally, we showed that MIF increased the phosphorylation of extracellular regulated protein kinases (ERK) 1/2 and focal adhesion kinase (FAK), which are associated with proliferation and migration. In conclusion, this study demonstrated that MIF overexpression promotes the proliferation and migration of ASMCs by upregulating the activity of the ERK1/2 and FAK signaling pathways in these cells, further indicating that inhibition of MIF may prove to be an effective strategy for treating asthma patients with airway remodeling.© 2017 International Federation for Cell Biology.
Sp1 transcription factor: a long-standing target in cancer chemotherapy
ACTL6A suppresses p21Cip1 tumor suppressor expression to maintain an aggressive mesothelioma cancer cell phenotype
Mesothelioma is a poor prognosis cancer of the mesothelial lining that develops in response to exposure to various agents including asbestos. Actin-Like Protein 6A (ACTL6A, BAF53a) is a SWI/SNF regulatory complex protein that is elevated in cancer cells and has been implicated as a driver of cancer cell survival and tumor formation. In the present study, we show that ACTL6A drives mesothelioma cancer cell proliferation, spheroid formation, invasion, and migration, and that these activities are markedly attenuated by ACTL6A knockdown. ACTL6A expression reduces the levels of the p21 cyclin-dependent kinase inhibitor and tumor suppressor protein. DNA binding studies show that ACTL6A interacts with Sp1 and p53 binding DNA response elements in the p21 gene promoter and that this is associated with reduced p21 promoter activity and p21 mRNA and protein levels. Moreover, ACTL6A suppression of p21 expression is required for maintenance of the aggressive mesothelioma cancer cell phenotype suggesting that p21 is a mediator of ACTL6A action. p53, a known inducer of p21 expression, is involved ACTL6A in regulation of p21 in some but not all mesothelioma cells. In addition, ACTL6A knockout markedly reduces tumor formation and this is associated with elevated tumor levels of p21. These findings suggest that ACTL6A suppresses p21 promoter activity to reduce p21 protein as a mechanism to maintain the aggressive mesothelioma cell phenotype.© 2021. The Author(s).
Macrophage migration inhibitory factor confers resistance to senescence through CD74-dependent AMPK-FOXO3a signaling in mesenchymal stem cells
Exosomal LncRNA-NEAT1 derived from MIF-treated mesenchymal stem cells protected against doxorubicin-induced cardiac senescence through sponging miR-221-3p
The chemotherapy drug doxorubicin (Dox) is widely used for treating a variety of cancers. However, its high cardiotoxicity hampered its clinical use. Exosomes derived from stem cells showed a therapeutic effect against Dox-induced cardiomyopathy (DIC). Previous studies reported that exosomes derived from mesenchymal stem cells (MSCs) pretreated with macrophage migration inhibitory factor (MIF) (exosome) showed a cardioprotective effect through modulating long noncoding RNAs/microRNAs (lncRNAs/miRs). This study aimed to investigate the role of exosome in the treatment of DIC.Exosomes were isolated from control MSCs (exosome) and MIF-pretreated MSCs (exosome). Regulatory lncRNAs activated by MIF pretreatment were explored using genomics approaches. Fluorescence-labeled exosomes were tracked in vitro by fluorescence imaging. In vivo and in vitro, miR-221-3p mimic transfection enforced miR-221-3p overexpression, and senescence-associated β-galactosidase assay was applied to test cellular senescence. Exosomal delivering LncRNA-NEAT1 induced therapeutic effect in vivo was confirmed by echocardiography. It demonstrated that exosomes recovered the cardiac function and exerted the anti-senescent effect through LncRNA-NEAT1 transfer against Dox. TargetScan and luciferase assay showed that miR-221-3p targeted the Sirt2 3'-untranslated region. Silencing LncRNA-NEAT1 in MSCs, miR-221-3p overexpression or Sirt2 silencing in cardiomyocytes decreased the exosome-induced anti-senescent effect against Dox.The results indicated exosome serving as a promising anti-senescent effector against Dox-induced cardiotoxicity through LncRNA-NEAT1 transfer, thus inhibiting miR-221-3p and leading to Sirt2 activation. The study proposed that exosome might have the potential to serve as a cardioprotective therapeutic agent during cancer chemotherapy.
Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart
Elderly patients are more sensitive than younger patients to myocardial ischemia, which results in higher mortality. We investigated how aging affects the cardioprotective AMP-activated protein kinase (AMPK) signaling pathway.Ischemic AMPK activation was impaired in aged compared with young murine hearts. The expression and secretion of the AMPK upstream regulator, macrophage migration inhibitory factor (MIF), were lower in aged compared with young adult hearts. Additionally, the levels of hypoxia-inducible factor 1alpha, a known transcriptional activator of MIF, were reduced in aged compared with young hearts. Ischemia-induced AMPK activation in MIF knockout mice was blunted, leading to greater contractile dysfunction in MIF-deficient than in wild-type hearts. Furthermore, intramyocardial injection of adenovirus encoding MIF in aged mice increased MIF expression and ischemic AMPK activation and reduced infarct size.An impaired MIF-AMPK activation response in senescence thus may be attributed to an aging-associated defect in hypoxia-inducible factor 1alpha, the transcription factor for MIF. In the clinical setting, impaired cardiac hypoxia-inducible factor 1alpha activation and consequent reduced MIF expression may play an important role in the increased susceptibility to myocardial ischemia observed in older cardiac patients.
Theories of aging: an ever-evolving field
Senescence has been the focus of research for many centuries. Despite significant progress in extending average human life expectancy, the process of aging remains largely elusive and, unfortunately, inevitable. In this review, we attempted to summarize the current theories of aging and the approaches to understanding it.
Nrf2: INrf 2 (Keap1) signaling in oxidative stress
Nrf2:INrf2 (Keap1) are cellular sensors of chemical- and radiation-induced oxidative and electrophilic stress. Nrf2 is a nuclear transcription factor that controls the expression and coordinated induction of a battery of defensive genes encoding detoxifying enzymes and antioxidant proteins. This is a mechanism of critical importance for cellular protection and cell survival. Nrf2 is retained in the cytoplasm by an inhibitor, INrf2 which functions as an adapter for Cul3/Rbx1-mediated degradation of Nrf2. In response to oxidative/electrophilic stress, Nrf2 is switched on and then off by distinct early and delayed mechanisms. Oxidative/electrophilic modification of INrf2 cysteine 151 and/or protein kinase C phosphorylation of Nrf2 serine 40 results in the escape or release of Nrf2 from INrf2. Nrf2 is stabilized and translocates to the nucleus, forms heterodimers with unknown proteins, and binds the antioxidant response element, which leads to coordinated activation of gene expression. It takes less than 15 min from the time of exposure to switch on nuclear import of Nrf2. This is followed by activation of a delayed mechanism that controls the switching off of Nrf2 activation of gene expression. GSK3beta phosphorylates Fyn at an unknown threonine residue(s), leading to the nuclear localization of Fyn. Fyn phosphorylates Nrf2 tyrosine 568, resulting in the nuclear export of Nrf2, binding with INrf2, and degradation of Nrf2. The switching on and off of Nrf2 protects cells against free radical damage, prevents apoptosis, and promotes cell survival.
BTZO-1, a cardioprotective agent, reveals that macrophage migration inhibitory factor regulates ARE-mediated gene expression
Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53:regulatory role in the innate immune response
Macrophage migration inhibitory factor provides cardioprotection during ischemia/reperfusion by reducing oxidative stress
Autophagy inhibition induces the secretion of macrophage migration inhibitory factor (MIF) with autocrine and paracrine effects on the promotion of malignancy in breast cancer
Breast cancer is the main cause of cancer-related death in women in the world. Because autophagy is a known survival pathway for cancer cells, its inhibition is currently being explored in clinical trials for treating several types of malignancies. In breast cancer, autophagy has been shown to be necessary for the survival of cancer cells from the triple negative subtype (TNBC), which has the worst prognosis among breast cancers and currently has limited therapeutic options. Autophagy has also been involved in the regulation of protein secretion and, of importance for this work, the inhibition of autophagy is known to promote the secretion of proinflammatory cytokines from distinct cell types. We found that the inhibition of autophagy in TNBC cell lines induced the secretion of the macrophage migration inhibitory factor (MIF), a pro-tumorigenic cytokine involved in breast cancer invasion and immunomodulation. MIF secretion was dependent on an increase in reactive oxygen species (ROS) induced by the inhibition of autophagy. Importantly, MIF secreted from autophagy-deficient cells increased the migration of cells not treated with autophagy inhibitors, indicating that autophagy inhibition in cancer cells promoted malignancy in neighboring cells through the release of secreted factors, and that a combinatorial approach should be evaluated for cancer therapy.
Chloroquine induces ROS-mediated macrophage migration inhibitory factor secretion and epithelial to mesenchymal transition in ER-positive breast cancer cell lines
Breast cancer (BC) is the leading cause of cancer-related death in women in the world. Since tumor cells employ autophagy as a survival pathway, it has been proposed that autophagy inhibition could be beneficial for cancer treatment. There are several onging clinical trials where autophagy is being inhibited (using chloroquine, CQ or hydroxychloroquine, HCQ) along with chemotherapy with promising results. However, there is also in vitro evidence in which autophagy inhibition can induce epithelial to mesenchymal transition (EMT) in cancer cells, indicating that, at least in some cases, this strategy could be detrimental for cancer patients. In this study, we found that the genetic inhibition of autophagy primed cells for EMT by inducing a decrease in E-cadherin protein levels, while CQ treatment decreased E-cadherin levels, induced morphological changes related to EMT, increased EMT-related transcription factor (EMT-TF) expression and migration in estrogen receptor positive (ER +) BC cell lines. Importantly, CQ treatment increased intracellular reactive oxygen species (ROS) which induced the secretion of macrophage migration inhibitory factor (MIF), a pro-inflammatory cytokine related to malignancy. Both ROS production and MIF secretion were responsible for the mesenchymal morphology and increased migratory capacity induced by CQ. Our results indicate that CQ treatment increased malignancy by inducing ROS production, MIF secretion and EMT and suggest that autophagy inhibition in ER + BC patients might have detrimental effects. Our data indicates that a careful selection of patients should be performed in order to determine who will benefit the most from autophagy inhibition with available pharmacological agents for the treatment of breast cancer.© 2021. The Author(s), under exclusive licence to Springer Science+Business Media, LLC, part of Springer Nature.
Hypoxia-inducible factor-1 (HIF-1)
Adaptation to low oxygen tension (hypoxia) in cells and tissues leads to the transcriptional induction of a series of genes that participate in angiogenesis, iron metabolism, glucose metabolism, and cell proliferation/survival. The primary factor mediating this response is the hypoxia-inducible factor-1 (HIF-1), an oxygen-sensitive transcriptional activator. HIF-1 consists of a constitutively expressed subunit HIF-1beta and an oxygen-regulated subunit HIF-1alpha (or its paralogs HIF-2alpha and HIF-3alpha). The stability and activity of the alpha subunit of HIF are regulated by its post-translational modifications such as hydroxylation, ubiquitination, acetylation, and phosphorylation. In normoxia, hydroxylation of two proline residues and acetylation of a lysine residue at the oxygen-dependent degradation domain (ODDD) of HIF-1alpha trigger its association with pVHL E3 ligase complex, leading to HIF-1alpha degradation via ubiquitin-proteasome pathway. In hypoxia, the HIF-1alpha subunit becomes stable and interacts with coactivators such as cAMP response element-binding protein binding protein/p300 and regulates the expression of target genes. Overexpression of HIF-1 has been found in various cancers, and targeting HIF-1 could represent a novel approach to cancer therapy.
Hypoxia stimulates the expression of macrophage migration inhibitory factor in human vascular smooth muscle cells via HIF-1alpha dependent pathway
Background: Hypoxia plays an important role in vascular remodeling and directly affects vascular smooth muscle cells (VSMC) functions. Macrophage migration inhibitory factor (MIF) is a well known proinflammatory factor, and recent evidence suggests an important role of MIF in the progression of atherosclerosis and restenosis. However, the potential link between hypoxia and MIF in VSMC has not been investigated. The current study was designed to test whether hypoxia could regulate MIF expression in human VSMC. The effect of modulating MIF expression on hypoxia-induced VSMC proliferation and migration was also investigated at the same time. Results: Expression of MIF mRNA and protein was up-regulated as early as 2 hours in cultured human VSMCs after exposed to moderate hypoxia condition (3% O(2)). The up-regulation of MIF expression appears to be dependent on hypoxia-inducible transcription factor-1 alpha(HIF-1 alpha) since knockdown of HIF-1 alpha inhibits the hypoxia induction of MIF gene and protein expression. The hypoxia induced expression of MIF was attenuated by antioxidant treatment as well as by inhibition of extracellular signal-regulated kinase (ERK). Under moderate hypoxia conditions (3% O(2)), both cell proliferation and cell migration were increased in VSMC cells. Blocking the MIF by specific small interference RNA to MIF (MIF-shRNA) resulted in the suppression of proliferation and migration of VSMCs. Conclusion: Our results demonstrated that in VSMCs, hypoxia increased MIF gene expression and protein production. The hypoxia-induced HIF-1 alpha activation, reactive oxygen species (ROS) generation and ERK activation might be involved in this response. Both MIF and HIF-1 alpha mediated the hypoxia response of vascular smooth muscle cells, including cell migration and proliferation.
HIF-1α-regulated MIF activation and Nox2-dependent ROS generation promote Leishmania amazonensis killing by macrophages under hypoxia
Increasing attention is given to the finding that macrophages under hypoxia are capable of controlling infection by the intracellular protozoan parasite Leishmania amazonensis. The hypoxia-inducible factor (HIF)-1α has been shown to play an essential role in this enhanced innate immune response. Our study aimed to explore the HIF-1α-dependent mechanisms leading to reduced survival of the parasites residing in macrophages under low oxygen conditions. Hypoxia triggered (P < 0.01) NADPH oxidase 2 (Nox2) expression and reactive oxygen species (ROS) production in J774 macrophages upon 24-h infection with L. amazonensis. Furthermore, increased (P < 0.01) expression levels of HIF-1α and macrophage migration inhibitory factor (MIF) were detected in the infected cells grown at 3% oxygen tension. We found that either HIF-1α silencing, Nox2 inhibition or MIF antagonism caused a significant (P < 0.05) reversal of the improved leishmanicidal activity displayed by the hypoxic phagocytes. Taken together, our current results suggest that, under conditions of limited availability of oxygen, activation of the HIF-1α/MIF axis via Nox2/ROS induction promotes killing of L. amazonensis amastigotes by macrophages. Such protective mechanism might operate in L. amazonensis-infected tissues where low oxygen levels prevail.Copyright © 2018 Elsevier Inc. All rights reserved.
Macrophage migration inhibitory factor is regulated by HIF-1α and cAMP and promotes renal cyst cell proliferation in a macrophage-independent manner
Progressive cyst growth leads to decline of renal function in polycystic kidney disease. Macrophage migration inhibitory factor (MIF) was found to be upregulated in cyst-lining cells in a mouse model of polycystic kidney disease and to promote cyst growth. In addition, MIF can be secreted by tubular cells and may contribute to cyst growth in an autocrine manner. However, the underlying mechanisms leading to induction of MIF in cyst-lining cells remained elusive. Here, we demonstrate that hypoxia-inducible transcription factor (HIF) 1α upregulates MIF in cyst-lining cells in a tubule-specific PKD1 knockout mouse. Pharmacological stabilization of HIF-1α resulted in significant increase of MIF in cyst epithelial cells whereas tubule-specific knockout of HIF-1α prevented MIF upregulation. Identical regulation could be found for ABCA1, which has been shown to act as a transport protein for MIF. Furthermore, we show that MIF and ABCA1 are direct target genes of HIF-1α in human primary tubular cells. Next to HIF-1α and hypoxia, we found MIF being additionally regulated by cAMP which is a strong promotor of cyst growth. In line with these findings, HIF-1α- and cAMP-dependent in vitro cyst growth could be decreased by the MIF-inhibitor ISO-1 which resulted in reduced cyst cell proliferation. In conclusion, HIF-1α and cAMP regulate MIF in primary tubular cells and cyst-lining epithelial cells, and MIF promotes cyst growth in the absence of macrophages. In line with these findings, the MIF inhibitor ISO-1 attenuates HIF-1α- and cAMP-dependent in vitro cyst enlargement.
Macrophage migration inhibitory factor promotes Warburg effect via activation of the NF-κB/HIF-1α pathway in lung cancer
The role of macrophage migration inhibitory factor in adipose-derived stem cells under hypoxia
Background: Adipose-derived stem cells (ASCs) are multipotent mesenchymal stem cells characterized by their strong regenerative potential and low oxygen consumption. Macrophage migration inhibitory factor (MIF) is a multifunctional chemokine-like cytokine that is involved in tissue hypoxia. MIF is not only a major immunomodulator but also is highly expressed in adipose tissue such as subcutaneous adipose tissue of chronic non-healing wounds. In the present study, we investigated the effect of hypoxia on MIF in ASCs isolated from healthy versus inflamed adipose tissue.
Macrophage migration inhibitory factor activates hypoxia-inducible factor in a p53-dependent manner
Macrophage migration inhibitory factor: a regulator of innate immunity
For more than a quarter of a century, macrophage migration inhibitory factor (MIF) has been a mysterious cytokine. In recent years, MIF has assumed an important role as a pivotal regulator of innate immunity. MIF is an integral component of the host antimicrobial alarm system and stress response that promotes the pro-inflammatory functions of immune cells. A rapidly increasing amount of literature indicates that MIF is implicated in the pathogenesis of sepsis, and inflammatory and autoimmune diseases, suggesting that MIF-directed therapies might offer new treatment opportunities for human diseases in the future.
Macrophage migration inhibitory factor (MIF): a multifaceted cytokine regulated by genetic and physiological strategies
The dichotomic role of macrophage migration inhibitory factor in neurodegeneration
Macrophage migration inhibitory factor (MIF) is a pleiotropic cytokine expressed by different cell types and exerting multiple biological functions. It has been shown that MIF may be involved in several disorders, including neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS), Parkinson disease (PD), and Huntington disease (HD), that represent an unmet medical need. Therefore, further studies are needed to identify novel pathogenetic mechanisms that may translate into tailored therapeutic approaches so to improve patients’ survival and quality of life. Here, we reviewed the preclinical and clinical studies investigating the role of MIF in ALS, PD, and HD. The emerging results suggest that MIF might play a dichotomic role in these disorders, exerting a protective action in ALS, a pathogenetic action in HD, and a yet undefined and debated role in PD. The better understanding of the role of MIF in these diseases could allow its use as a novel diagnostic and therapeutic tool for the monitoring and treatment of the patients and for eventual biomarker-driven therapeutic approaches.
Macrophage migration inhibitory factor (MIF)-based therapeutic concepts in atherosclerosis and inflammation
Chemokines orchestrate leukocyte recruitment in atherosclerosis and their blockade is a promising anti-atherosclerotic strategy, but few chemokine-based approaches have advanced into clinical trials, in part owing to the complexity and redundancy of the chemokine network. Macrophage migration inhibitory factor (MIF) is a pivotal mediator of atherosclerotic lesion formation. It has been characterized as an inflammatory cytokine and atypical chemokine that promotes atherogenic leukocyte recruitment and lesional inflammation through interactions with the chemokine receptors CXCR2 and CXCR4, but also exhibits phase-specific CD74-mediated cardioprotective activity. The unique structural properties of MIF and its homologue MIF-2/D-DT offer intriguing therapeutic opportunities including small molecule-, antibody- and peptide-based approaches that may hold promise as inhibitors of atherosclerosis, while sparing tissue-protective classical chemokine pathways. In this review, we summarize the pros and cons of anti-MIF protein strategies and discuss their molecular characteristics and receptor specificities with a focus on cardiovascular disease.Georg Thieme Verlag KG Stuttgart · New York.
Role of macrophage migration inhibitory factor in age-related lung disease
The prevalence of many common respiratory disorders, including pneumonia, chronic obstructive lung disease, pulmonary fibrosis, and lung cancer, increases with age. Little is known of the host factors that may predispose individuals to such diseases. Macrophage migration inhibitory factor (MIF) is a potent upstream regulator of the immune system. MIF is encoded by variant alleles that occur commonly in the population. In addition to its role as a proinflammatory cytokine, a growing body of literature demonstrates that MIF influences diverse molecular processes important for the maintenance of cellular homeostasis and may influence the incidence or clinical manifestations of a variety of chronic lung diseases. This review highlights the biological properties of MIF and its implication in age-related lung disease.
Macrophage migration inhibitory factor (MIF): biological activities and relation with cancer
Macrophage migration inhibitory factor mediates neuroprotective effects by regulating inflammation, apoptosis and autophagy in Parkinson’s disease
Upregulation of MIF as a defense mechanism and a biomarker of Alzheimer’s disease
Critical role of cysteine residue 81 of macrophage migration inhibitory factor (MIF) in MIF-induced inhibition of p53 activity
Macrophage migration inhibitory factor (MIF) is a potent modulator of the p53 signaling pathway, but the molecular mechanisms of the effect of MIF on p53 function have so far remained unclear. Here we show that MIF physically interacts with the p53 tumor suppressor in vitro and in vivo. This association was significantly reduced by a C81S mutation but not C57S or C60S mutations, suggesting that Cys(81) is essential for the in vivo association between MIF and p53. This association also depended on Cys(242) (and, to some extent, on Cys(238)) within the central DNA binding domain of p53. Ectopic expression of MIF, but not MIF(C81S), inhibited p53-mediated transcriptional activation in a dose-dependent manner. Conversely, knockdown of endogenous MIF stimulated p53-mediated transcription. MIF inhibited p53-induced apoptosis and cell cycle arrest, whereas the MIF(C81S) mutant, which is unable to physically associate with p53, had no effect. Consistent with these findings, confocal microscopy showed that MIF prevented p53 translocation from the cytoplasm to the nucleus. We also demonstrated that MIF suppresses p53 activity by stabilizing the physical association between p53 and Mdm2. These results suggest that MIF physically associates with p53 and negatively regulates p53 function.
Macrophage migration inhibitory factor regulating the expression of VEGF-C through MAPK signal pathways in breast cancer MCF-7 cell
MIF/CXCR4 signaling axis contributes to survival, invasion, and drug resistance of metastatic neuroblastoma cells in the bone marrow microenvironment
The bone marrow (BM) is the most common site of dissemination in patients with aggressive, metastatic neuroblastoma (NB). However, the molecular mechanisms underlying the aggressive behavior of NB cells in the BM niche are still greatly unknown. In the present study, we explored biological mechanisms that play a critical role in NB cell survival and progression in the BM and investigated potential therapeutic targets.Patient-derived bone marrow (BM) primary cultures were generated using fresh BM aspirates obtained from NB patients. NB cell lines were cultured in the presence of BM conditioned media containing cell-secreted factors, and under low oxygen levels (1% O) to mimic specific features of the BM microenvironment of high-risk NB patients. The BM niche was explored using cytokine profiling assays, cell migration-invasion and viability assays, flow cytometry and analysis of RNA-sequencing data. Selective pharmacological inhibition of factors identified as potential mediators of NB progression within the BM niche was performed in vitro and in vivo.We identified macrophage migration inhibitory factor (MIF) as a key inflammatory cytokine involved in BM infiltration. Cytokine profiling and RNA-sequencing data analysis revealed NB cells as the main source of MIF in the BM, suggesting a potential role of MIF in tumor invasion. Exposure of NB cells to BM-conditions increased NB cell-surface expression of the MIF receptor CXCR4, which was associated with increased cell viability, enhanced migration-invasion, and activation of PI3K/AKT and MAPK/ERK signaling pathways. Moreover, subcutaneous co-injection of NB and BM cells enhanced tumor engraftment in mice. MIF inhibition with 4-IPP impaired in vitro NB aggressiveness, and improved drug response while delayed NB growth, improving survival of the NB xenograft model.Our findings suggest that BM infiltration by NB cells may be mediated, in part, by MIF-CXCR4 signaling. We demonstrate the antitumor efficacy of MIF targeting in vitro and in vivo that could represent a novel therapeutic target for patients with disseminated high-risk NB.© 2022. The Author(s).
MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment
The cytokine macrophage migration inhibitory factor (MIF) plays a critical role in inflammatory diseases and atherogenesis. We identify the chemokine receptors CXCR2 and CXCR4 as functional receptors for MIF. MIF triggered G(alphai)- and integrin-dependent arrest and chemotaxis of monocytes and T cells, rapid integrin activation and calcium influx through CXCR2 or CXCR4. MIF competed with cognate ligands for CXCR4 and CXCR2 binding, and directly bound to CXCR2. CXCR2 and CD74 formed a receptor complex, and monocyte arrest elicited by MIF in inflamed or atherosclerotic arteries involved both CXCR2 and CD74. In vivo, Mif deficiency impaired monocyte adhesion to the arterial wall in atherosclerosis-prone mice, and MIF-induced leukocyte recruitment required Il8rb (which encodes Cxcr2). Blockade of Mif but not of canonical ligands of Cxcr2 or Cxcr4 in mice with advanced atherosclerosis led to plaque regression and reduced monocyte and T-cell content in plaques. By activating both CXCR2 and CXCR4, MIF displays chemokine-like functions and acts as a major regulator of inflammatory cell recruitment and atherogenesis. Targeting MIF in individuals with manifest atherosclerosis can potentially be used to treat this condition.
Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart
Macrophage migration inhibitory factor is a novel determinant of cigarette smoke-induced lung damage
Cigarette smoke (CS) is the most common cause of chronic obstructive pulmonary diseases (COPD), including emphysema. CS exposure impacts all cell types within the airways and lung parenchyma, causing alveolar tissue destruction through four mechanisms: (1) oxidative stress; (2) inflammation; (3) protease-induced degradation of the extracellular matrix; and (4) enhanced alveolar epithelial and endothelial cell (EC) apoptosis. Studies in human pulmonary ECs demonstrate that macrophage migration inhibitory factor (MIF) antagonizes CS-induced apoptosis. Here, we used human microvascular ECs, an animal model of emphysema (mice challenged with chronic CS), and patient serum samples to address both the capacity of CS to alter MIF expression and the effects of MIF on disease severity. We demonstrate significantly reduced serum MIF levels in patients with COPD. In the murine model, chronic CS exposure resulted in decreased MIF mRNA and protein expression in the intact lung. MIF deficiency (Mif(-/-)) potentiated the toxicity of CS exposure in vivo via increased apoptosis of ECs, resulting in enhanced CS-induced tissue remodeling. This was linked to MIF's capacity to protect against double-stranded DNA damage and suppress p53 expression. Taken together, MIF appears to antagonize CS-induced toxicity in the lung and resultant emphysematous tissue remodeling by suppressing EC DNA damage and controlling p53-mediated apoptosis, highlighting a critical role of MIF in EC homeostasis within the lung.
Role of migratory inhibition factor in age-related susceptibility to radiation lung injury via NF-E2-related factor-2 and antioxidant regulation
Microvascular injury and increased vascular leakage are prominent features of radiation-induced lung injury (RILI), and often follow cancer-associated thoracic irradiation. Our previous studies demonstrated that polymorphisms in the gene (MIF) encoding macrophage migratory inhibition factor (MIF), a multifunctional pleiotropic cytokine, confer susceptibility to acute inflammatory lung injury and increased vascular permeability, particularly in senescent mice. In this study, we exposed wild-type and genetically engineered mif(-/-) mice to 20 Gy single-fraction thoracic radiation to investigate the age-related role of MIF in murine RILI (mice were aged 8 wk, 8 mo, or 16 mo). Relative to 8-week-old mice, decreased MIF was observed in bronchoalveolar lavage fluid and lung tissue of 8- to 16-month-old wild-type mice. In addition, radiated 8- to 16-month-old mif(-/-) mice exhibited significantly decreased bronchoalveolar lavage fluid total antioxidant concentrations with progressive age-related decreases in the nuclear expression of NF-E2-related factor-2 (Nrf2), a transcription factor involved in antioxidant gene up-regulation in response to reactive oxygen species. This was accompanied by decreases in both protein concentrations (NQO1, GCLC, and heme oxygenase-1) and mRNA concentrations (Gpx1, Prdx1, and Txn1) of Nrf2-influenced antioxidant gene targets. In addition, MIF-silenced (short, interfering RNA) human lung endothelial cells failed to express Nrf2 after oxidative (H2O2) challenge, an effect reversed by recombinant MIF administration. However, treatment with an antioxidant (glutathione reduced ester), but not an Nrf2 substrate (N-acetyl cysteine), protected aged mif(-/-) mice from RILI. These findings implicate an important role for MIF in radiation-induced changes in lung-cell antioxidant concentrations via Nrf2, and suggest that MIF may contribute to age-related susceptibility to thoracic radiation.
Role of macrophage migration inhibitory factor in age-related hearing loss
Hearing loss related to aging is the most common sensory disorder among elderly individuals. Macrophage migration inhibitory factor (MIF) is a multi-functional molecule. The aim of this study was to identify the role of MIF in the inner ear. MIF-deficient mice (MIF(-/-) mice) of BALB/c background and wild-type BALB/c mice were used in this study. Expression of MIF protein in the inner ear was examined by immunohistochemistry in wild-type mice (WT). The hearing function was assessed by the click-evoked auditory brainstem response in both MIF(-/-) mice and WT at 1, 3, 6, 9, 12, and 18months of age. Morphological examination of the cochlea was also performed using scanning electron microscopy and light microscopy. MIF was observed in the spiral ligament, stria vascularis, Reissner's membrane, spiral ganglion cells (SGCs), saccular macula, and membranous labyrinth. The MIF(-/-) mice had a significant hearing loss as compared with the WT at 9, 12, and 18months of age. In the MIF(-/-) mice, scanning electron microscopy showed that the outer cochlear hair cells were affected, but that the inner cochlear hair cells were relatively well preserved. The number of SGCs was lower in the MIF(-/-) mice. MIF was strongly expressed in the mouse inner ear. Older MIF(-/-) mice showed accelerated age-related hearing loss and morphological inner ear abnormalities. These findings suggest that MIF plays an important role in the inner ear of mice. Copyright © 2014 IBRO. Published by Elsevier Ltd. All rights reserved.
Reduced osteoarthritis severity in aged mice with deletion of macrophage migration inhibitory factor
Macrophage migration inhibitory factor may play a protective role in osteoarthritis
Estrogen modulates cutaneous wound healing by downregulating macrophage migration inhibitory factor
Phase I study of imalumab (BAX69), a fully human recombinant antioxidized macrophage migration inhibitory factor antibody in advanced solid tumours
Human anti-macrophage migration inhibitory factor antibodies inhibit growth of human prostate cancer cells in vitro and in vivo
Macrophage migration inhibitory factor (MIF) is a proinflammatory cytokine, originally discovered for its eponymous effect and now known for pleiotropic biologic properties in immunology and oncology. Circulating MIF levels are elevated in several types of human cancer including prostate cancer. MIF is released presumably by both stromal and tumor cells and enhances malignant growth and metastasis by diverse mechanisms, such as stimulating tumor cell proliferation, suppressing apoptotic death, facilitating invasion of the extracellular matrix, and promoting angiogenesis. Recently described fully human anti-MIF antibodies were tested in vitro and in vivo for their ability to influence growth rate and invasion of the human PC3 prostate cancer cell line. In vitro, the selected candidate antibodies BaxG03, BaxB01, and BaxM159 reduced cell growth and viability by inhibiting MIF-induced phosphorylation of the central kinases p44/42 mitogen-activated protein kinase [extracellular signal-regulated kinase-1 and -2 (ERK1/2)] and protein kinase B (AKT). Incubation of cells in the presence of the antibodies also promoted activation of caspase-3/7. The antibodies furthermore inhibited MIF-promoted invasion and chemotaxis as transmigration through Matrigel along a MIF gradient was impaired. In vivo, pharmacokinetic parameters (half-life, volume of distribution, and bioavailability) of the antibodies were determined and a proof-of-concept was obtained in a PC3-xenograft mouse model. Treatment with human anti-MIF antibodies blunted xenograft tumor growth in a dose-dependent manner. We therefore conclude that the anti-MIF antibodies described neutralize some of the key tumor-promoting activities of MIF and thus limit tumor growth in vivo.
Combining milatuzumab with bortezomib, doxorubicin, or dexamethasone improves responses in multiple myeloma cell lines
Purpose: The humanized anti-CD74 monoclonal antibody, milatuzumab, is in clinical evaluation for the therapy of multiple myeloma (MM). The ability of milatuzumab to increase the efficacy of bortezomib, doxorubicin, and dexamethasone was examined in three human CD74+ MM cell lines, CAG, KMS11, KMS12-PE, and one CD74-MM cell line, OPM-2.
Dissecting the role of the CXCL12/CXCR4 axis in acute myeloid leukaemia
Acute myeloid leukaemia (AML) is the most common adult acute leukaemia with the lowest survival rate. It is characterised by a build-up of immature myeloid cells anchored in the protective niche of the bone marrow (BM) microenvironment. The CXCL12/CXCR4 axis is central to the pathogenesis of AML as it has fundamental control over AML cell adhesion into the protective BM niche, adaptation to the hypoxic environment, cellular migration and survival. High levels of CXCR4 expression are associated with poor relapse-free and overall survival. The CXCR4 ligand, CXCL12 (SDF-1), is expressed by multiple cells types in the BM, facilitating the adhesion and survival of the malignant clone. Blocking the CXCL12/CXCR4 axis is an attractive therapeutic strategy providing a 'multi-hit' therapy that both prevents essential survival signals and releases the AML cells from the BM into the circulation. Once out of the protective niche of the BM they would be more susceptible to destruction by conventional chemotherapeutic drugs. In this review, we disentangle the diverse roles of the CXCL12/CXCR4 axis in AML. We then describe multiple CXCR4 inhibitors, including small molecules, peptides, or monoclonal antibodies, which have been developed to date and their progress in pre-clinical and clinical trials. Finally, the review leads us to the conclusion that there is a need for further investigation into the development of a 'multi-hit' therapy that targets several signalling pathways related to AML cell adhesion and maintenance in the BM.© 2020 British Society for Haematology and John Wiley & Sons Ltd.
A phase Ib/II trial of the first-in-class anti-CXCR4 antibody ulocuplumab in combination with lenalidomide or bortezomib plus dexamethasone in relapsed multiple myeloma
Ulocuplumab (BMS-936564) is a first-in-class fully human IgG4 monoclonal anti-CXCR4 antibody that inhibits the binding of CXCR4 to CXCL12.
Phase1 study of ibrutinib and the CXCR4 antagonist ulocuplumab in CXCR4-mutated Waldenström macroglobulinemia
Ulocuplumab (BMS-936564/MDX1338): a fully human anti-CXCR4 antibody induces cell death in chronic lymphocytic leukemia mediated through a reactive oxygen species-dependent pathway
The CXCR4 receptor (Chemokine C-X-C motif receptor 4) is highly expressed in different hematological malignancies including chronic lymphocytic leukemia (CLL). The CXCR4 ligand (CXCL12) stimulates CXCR4 promoting cell survival and proliferation, and may contribute to the tropism of leukemia cells towards lymphoid tissues. Therefore, strategies targeting CXCR4 may constitute an effective therapeutic approach for CLL. To address that question, we studied the effect of Ulocuplumab (BMS-936564), a fully human IgG4 anti-CXCR4 antibody, using a stroma--CLL cells co-culture model. We found that Ulocuplumab (BMS-936564) inhibited CXCL12 mediated CXCR4 activation-migration of CLL cells at nanomolar concentrations. This effect was comparable to AMD3100 (Plerixafor--Mozobil), a small molecule CXCR4 inhibitor. However, Ulocuplumab (BMS-936564) but not AMD3100 induced apoptosis in CLL at nanomolar concentrations in the presence or absence of stromal cell support. This pro-apoptotic effect was independent of CLL high-risk prognostic markers, was associated with production of reactive oxygen species and did not require caspase activation. Overall, these findings are evidence that Ulocuplumab (BMS-936564) has biological activity in CLL, highlight the relevance of the CXCR4-CXCL12 pathway as a therapeutic target in CLL, and provide biological rationale for ongoing clinical trials in CLL and other hematological malignancies.
Characterization and application of two novel monoclonal antibodies against human CXCR4: cell proliferation and migration regulation for glioma cell line in vitro by CXCR4/SDF-1alpha signal
Small-molecule inhibitors of macrophage migration inhibitory factor (MIF) as an emerging class of therapeutics for immune disorders
Macrophage migration inhibitory factor (MIF) is an important cytokine for which an increasing number of functions is being described in the pathogenesis of inflammation and cancer. Nevertheless, the availability of potent and druglike MIF inhibitors that are well-characterized in relevant disease models remains limited. Development of highly potent and selective small-molecule MIF inhibitors and validation of their use in relevant disease models will advance drug discovery. In this review, we provide an overview of recent advances in the identification of MIF as a pharmacological target in the pathogenesis of inflammatory diseases and cancer. We also give an overview of the current developments in the discovery and design of small-molecule MIF inhibitors and define future aims in this field.Copyright © 2018 Elsevier Ltd. All rights reserved.
MIF inhibitor ISO-1 alleviates severe acute pancreatitis-associated acute kidney injury by suppressing the NLRP 3 inflammasome signaling pathway
MIF inhibitor, ISO-1, attenuates human pancreatic cancer cell proliferation, migration and invasion in vitro, and suppresses xenograft tumour growth in vivo
This study sought to investigate the biological effects of specific MIF inhibitor, ISO-1, on the proliferation, migration and invasion of PANC-1 human pancreatic cells in vitro, and on tumour growth in a xenograft tumour model in vivo. The effect of ISO-1 on PANC-1 cell proliferation was examined using CCK-8 cell proliferation assay. The effect of ISO-1 on collective cell migration and recolonization of PANC-1 cells was evaluated using the cell-wound closure migration assay. The effect of ISO-1 on the migration and invasion of individual PANC-1 cells in a 3-dimensional environment in response to a chemo-attractant was investigated through the use of Transwell migration/invasion assays. Quantitative real time PCR and western blot analyses were employed to investigate the effects of ISO-1 on MIF, NF-κB p65 and TNF-α mRNA and protein expression respectively. Finally, a xenograft tumor model in BALB/c nude mice were used to assess the in vivo effects of ISO-1 on PANC-1-induced tumor growth. We found high expression of MIF in pancreatic cancer tissues. We demonstrated that ISO-1 exerts anti-cancer effects on PANC-1 cell proliferation, migration and invasion in vitro, and inhibited PANC-1 cell-induced tumour growth in xenograft mice in vivo. Our data suggests that ISO-1 and its derivative may have potential therapeutic applications in pancreatic cancer.
Macrophage migration inhibitory factor (MIF) inhibitor 4-IPP suppresses osteoclast formation and promotes osteoblast differentiation through the inhibition of the NF-κB signaling pathway
Repertaxin, an inhibitor of the chemokine receptors CXCR1 and CXCR2, inhibits malignant behavior of human gastric cancer MKN45 cells in vitro and in vivo and enhances efficacy of 5-fluorouracil
A novel allosteric inhibitor of macrophage migration inhibitory factor (MIF)
Phase I study of the anti-CD 74 monoclonal antibody milatuzumab (hLL1) in patients with previously treated B-cell lymphomas
The development of nanobodies for therapeutic applications
Nanobodies from camelid mice and llamas neutralize SARS-CoV-2 variants
Since the start of the COVID-19 pandemic, SARS-CoV-2 has caused millions of deaths worldwide. Although a number of vaccines have been deployed, the continual evolution of the receptor-binding domain (RBD) of the virus has challenged their efficacy. In particular, the emerging variants B.1.1.7, B.1.351 and P.1 (first detected in the UK, South Africa and Brazil, respectively) have compromised the efficacy of sera from patients who have recovered from COVID-19 and immunotherapies that have received emergency use authorization1–3. One potential alternative to avert viral escape is the use of camelid VHHs (variable heavy chain domains of heavy chain antibody (also known as nanobodies)), which can recognize epitopes that are often inaccessible to conventional antibodies4. Here, we isolate anti-RBD nanobodies from llamas and from mice that we engineered to produce VHHs cloned from alpacas, dromedaries and Bactrian camels. We identified two groups of highly neutralizing nanobodies. Group 1 circumvents antigenic drift by recognizing an RBD region that is highly conserved in coronaviruses but rarely targeted by human antibodies. Group 2 is almost exclusively focused to the RBD–ACE2 interface and does not neutralize SARS-CoV-2 variants that carry E484K or N501Y substitutions. However, nanobodies in group 2 retain full neutralization activity against these variants when expressed as homotrimers, and—to our knowledge—rival the most potent antibodies against SARS-CoV-2 that have been produced to date. These findings suggest that multivalent nanobodies overcome SARS-CoV-2 mutations through two separate mechanisms: enhanced avidity for the ACE2-binding domain and recognition of conserved epitopes that are largely inaccessible to human antibodies. Therefore, although new SARS-CoV-2 mutants will continue to emerge, nanobodies represent promising tools to prevent COVID-19 mortality when vaccines are compromised.
Versatile and multivalent nanobodies efficiently neutralize SARS-CoV-2
Cost-effective, efficacious therapeutics are urgently needed to combat the COVID-19 pandemic. In this study, we used camelid immunization and proteomics to identify a large repertoire of highly potent neutralizing nanobodies (Nbs) to the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein receptor binding domain (RBD). We discovered Nbs with picomolar to femtomolar affinities that inhibit viral infection at concentrations below the nanograms-per-milliliter level, and we determined a structure of one of the most potent Nbs in complex with the RBD. Structural proteomics and integrative modeling revealed multiple distinct and nonoverlapping epitopes and indicated an array of potential neutralization mechanisms. We bioengineered multivalent Nb constructs that achieved ultrahigh neutralization potency (half-maximal inhibitory concentration as low as 0.058 ng/ml) and may prevent mutational escape. These thermostable Nbs can be rapidly produced in bulk from microbes and resist lyophilization and aerosolization.Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Novel half-life extended anti-MIF nanobodies protect against endotoxic shock

{kind=link}
{kind=link}
{kind=link}
{kind=link}